Nitric oxide (NO) is a short-lived free radical, generated from L-arginine by NO synthase (NOS) in an enzymatic reaction. It is diffusible across the biological membrane and has such diverse roles as an endothelium derived relaxing factor, a neuromodulator, and an immunological mediator in the cardiovascular, nervous, and immune systems.1,2 In the eye, NO is a physiological regulator as well as a pathological mediator not only in the vascular system but also in other tissues including the retina and the ocular surface.3
It has been proposed that the pathogenesis of primary open-angle glaucoma may be related to the alteration in the trabecular meshwork (TM), the ocular tissue believed to be responsible for the majority of aqueous humor outflow resistance. TM cells perform a variety of activities in maintaining the normal function of the aqueous outflow pathway.
The activity of nitric oxide synthase has been shown in the TM of bovine, porcine and human eyes.4-6 Since TM possess a contractile properties and the main isoform of NOS in the ocular outflow pathway is known to an eNOS,4 NO can affect outflow facility by relaxation of TM. It is also known that NO is involved in the regulation of trabecular outflow and external administration of NO results in decreased IOP.7,8 Alteration of NOS activity is reported in the glaucomatous eyes9 and basal NO production is enhanced by hydraulic pressure in cultured human TM cells.10
Endothelial dysfunction is characterized by an inability of endothelial cells to produce adequate amount of bioactive NO and hyperglycemia contributes to endothelial dysfunction and decreased NO bioavailability11,12 but the mechanisms remain uncharacterized. It has been proposed that high glucose inhibits basal eNOS expression, activity, and NO production in cultured endothelial cells.13,14 Insulin is a direct-acting vasodilator in intact vessels and has been demonstrated to stimulate NO production in cultured endothelial cells.15-17
Taken together, there is a possibility that insulin can affect on the formation of NO in TM cells. The present study was designed to investigate whether insulin affect NO production in human TM cells. Additionally the enzymatic synthetic pathway for tetrahydrobiopterin (BH4) synthesis, an essential cofactor for NO synthesis, was also evaluated.
Materials and Methods
Cell culture
TM cell cultures were established from enucleated human eyes obtained from an eye bank and transported on ice within 2 hr after exsanguinations as previously described.18,19 Briefly, TM tissues were excised by dissecting a continuous strand of tissue between the line of Schwalbe and the scleral spur, using a curette to separate a continuous strand of TM tissue away from the scleral spur, ciliary muscle and Schwalbe's line. The excised TM tissues were placed in the sterile laminin-coated culture dish with DMEM medium containing 15% fetal bovine serum, 2 mM glutamine, 50 ┬Ąg/ml gentamicin and 2.5 mg/ml fungizone and left undisturbed for 3 to 5 days in a 37Ōäā incubator with a 5% CO2 atmosphere. After identifying initial cell growth, the explants were removed and the cultures were maintained with a medium containing 10% fetal bovine serum. Passages three through five were used for experiments.
Experimental treatment
With approaching confluency, the cultures were trypsinized and inoculated into 24 well culture plates (1├Ś106 cells/well). After allowing 24 hours for attachment, 1, 10, 100, and 1000 nM of dexamethasone (Sigma, St. Louis, MO, USA) and 1, 10, and 100 ┬Ąg/ml of insulin (Sigma, St. Louis, MO, USA) were added to the media with or without NOS inhibitor, 0.5 mM NŽē-Nitro-L-arginine methyl ester (L-NAME: Sigma, St. Louis, MO, USA). In addition, 10 ┬ĄM dexamethasone, 5 mM diaminopyrimidinone, 100 ┬ĄM L-ascorbic acid, 100 ┬ĄM sepiapterin, or 10 ┬ĄM methotrexate were added to the medium respectively with co-exposure of insulin to evaluate enzymatic synthetic pathway of BH4 synthesis. The cultures were replaced to the incubator and incubated for 72 hours.
Cell viability
Cell viability was determined by a rapid colorimetric assay using 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT: Sigma, St Louis, MO, USA). The MTT assay is based on the ability of the tetrazolium salt MTT to detect living but not dead cells. The signal generated, optical density (OD) is directly proportional to the number of cells.20,21 After adding 100 ┬Ąl of an MTT stock solution (5 mg MTT/ml PBS) to each well, the media was completely removed from the well after a 4-hr incubation at 37Ōäā. Then 0.5 ml of dimethyl sulfoxide (DMSO: Sigma, St Louis, MO, USA) was added to each well and 100 ┬Ąl of the solution from each well was transferred to a 96-well plate and read at 570 nm on a multi-well scanning spectrophotometer. It was noted that a minimum 10 min exposure to DMSO was required to dissolve the MTT formazan crystals after which the absorbance remained constant for 20 min. Thus, in all experiments spectrophotometric readings were taken 15 min after the addition of DMSO.
Measurement of NO production
Nitrite's concentrations in the media were measured by Griess reaction.22 Briefly, media samples were collected from each wells following appropriate treatment and reacted with modified Griess reagent (Sigma, St Louis, MO, USA) by mixing equal volumes with each other at room temperature for 15 min. Optical density was then measured and read on a multi-well scanning spectrophotometer at 540 nm. The nitrite concentration was then determined from a comparison of absorbance with that of a standard solution of sodium nitrite in medium. The background absorbance measured using the medium alone, was subtracted from all values.
Statistical analysis
Data are expressed as mean┬▒standard error of the mean (mean┬▒SEM) corresponding to the number of wells analyzed. Experimental differences between control culture results and a single treatment group were evaluated using Student's t-test with P values less than 0.05 considered significant.
Results
Cell culture
The cultured human TM cells were similar in appearance to those human TM cells generated by other investigator and identification of TM cells was done by their characteristic morphology and growth pattern.18,19 Confluent cultures of TM cells grew as a monolayer of closely packed cells with branching cell bodies making multiple contacts between cells. The cells were flattened with rounded to moderately elongated bodies with each cell containing a centrally placed nucleus. Often the migrating TM cells showed ruffled borders as they detached from the main cell mass. Occasionally migrating cells formed satellite colonies of cells with identical morphology to the main cell mass.18,19 This cellular morphology was maintained at least for 5 passages.
Effect on cell viability
Insulin and dexamethsone did not affect significantly on the survival of cultured trabecular meshwork cells up to 100 ┬Ąg/ml and 1000 nM respectively. Other co-exposed chemical agents also did not affect cellular survival (data not shown). Thus the effect on NO production of the each chemical agent used at experiments does not influenced by cell numbers significantly.
Effect on NO production
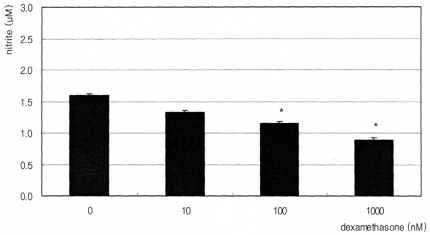
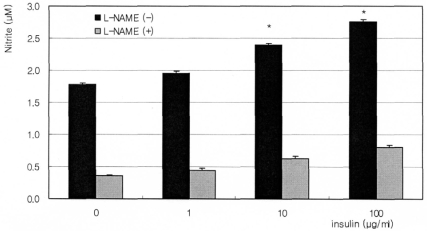
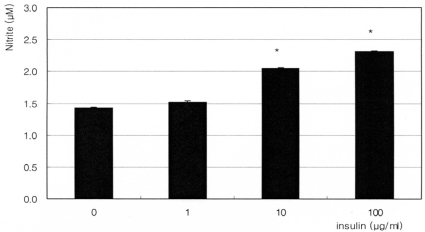
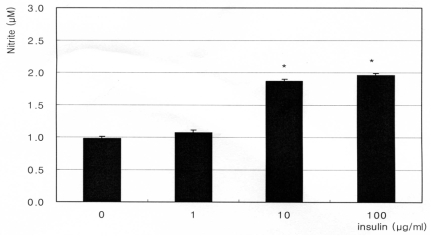
The synthetic glucocorticoid dexamethasone inhibited NO production significantly in a dose-dependent manner. Nitrite concentration was decreased from 1.60 ┬ĄM of non-exposed control down to 0.89 ┬ĄM of 1000 nM dexamethasone exposure (Fig. 1). On the contrary, insulin enhanced NO production significantly in a dose-dependent manner up to 2.76 ┬ĄM (Fig. 2). This insulin-induced enhanced NO production was inhibited by L-NAME, indicating NO was produced by TM cells per se. With co-exposure of insulin, 10 ┬ĄM dexamethasone did not affect to the insulin-induced increase of NO production (Fig. 3).
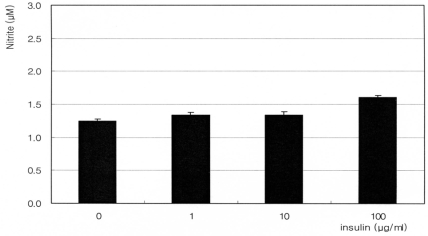
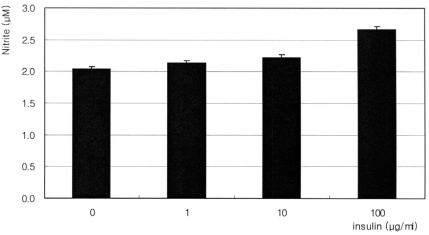
Co-exposure of 5 mM diaminopyrimidinone and 100 ┬ĄM sepiapterin, inhibitors of de novo synthetic pathway for BH4 synthesis, inhibited the insulin-induced increase of NO production (Fig. 4, 5). But co-exposure of 100 ┬ĄM L-ascorbic acid, a stimulator of de novo synthetic pathway for BH4 synthesis, increased NO production independent of the co-exposure of insulin (Fig. 6). Compared to single exposure of 0, 1, 10, and 100 ┬Ąg/ml, co-exposure of 100 ┬ĄM L-ascorbic further increased NO production 84%, 109.1%, 92.6%, and 103.3% respectively. Co-exposure of 10 ┬ĄM methotreaxate, an inhibitor of salvage pathway for BH4 synthesis, decreased overall NO production but did not affect to the insulin-induced increase of NO production (Fig. 7). Compared to single exposure of 0, 1, 10, and 100 ┬Ąg/ml, co-exposure of 10 ┬ĄM methotrexate further decreased NO production 56.9%, 54.6%, 77.8%, and 70.9% respectively. These findings suggest that insulin increases NO production via enhancing de novo synthetic pathway for BH4 synthesis in cultured TM cells.
Discussion
The main findings of this study are 1) dexamethasone inhibited NO production; 2) insulin enhanced NO production; and 3) enzymatic pathway of insulin-induced enhanced NO production is via stimulating de novo synthetic pathway for BH4 synthesis.
Since TM cells possess endothelial-like, smooth muscle cell-like activities, and NOS activities are expressed trabecular outflow pathway,4 NO could affect on the outflow facility by regulating the contractility TM. The concentration of endothelin, one of the most potent vasoconstrictors, which counteracts with NO,23 is increased in patients with progressive open angle glaucoma.24 In addition, NO possesses antiplatelet aggregatory activity and decreases leukocyte adhesion.1,3 Several studies have shown that NO is involved in the regulation of intraocular pressure and suggest that NO could increase the trabecular outflow by its innate dilating activity, thus result in the relaxation of TM and decrease of intraocular pressure. Recent findings have demonstrated that NO can serve as an ocular hypotensive agent.7,25-29 The NO/guanylate cyclase system plays a role in aqueous humor dynamics and in the regulation of intraocular pressure.30
In endothelial cells, high glucose inhibits NO production and result in oxidative stress subsequently.11-13 In the present study, a synthetic glucocorticoid dexamethasone decreased NO production in a dose-dependent manner in TM cells. Activity of eNOS can be modified in disease states, and in diabetes hyperglycemia inhibits eNOS as a result of a posttranslational modification at the protein kinase Akt regulatory site.14 Clinically such inhibition of NO production by glucose may be associated with increased intraocular pressure in steroid-induced glaucoma or diabetes (the concentration of glucose in the aqueous humor is higher in diabetes).31 Endothelin-1 expression was also induced by glucose32 and was increased in diabetes.33 On the contrary, insulin promotes vasodilatation by enhancing NO production in endothelial cells.15-17 As in the endothelial cells, insulin also increased NO production in TM cells in a dose-dependent manner in the present study, suggesting the beneficial effect of insulin on the regulation of trabecular outflow by relaxation activity of NO in TM.
The gasotransmitter NO is synthesized by oxidation of L-arginine and many cofactors are involved in the synthesis of NO. Among them, the pteridine cofactor BH4 has emerged as a critical determinant of NOS activity.34 BH4 is an essential cofactor for the proper flow of electrons to oxidize L-arginine, and NOS is the only heme-containing enzyme known to require a pterin cofactor for activity. Of significant interest, therefore, is that a reduced availability of BH4 may be a key factor in the etiology of the endothelial dysfunction. BH4 synthesis occurs via two distinct pathways: a de novo synthetic pathway which uses GTP as a precursor and a salvage pathway for preexisting dihydropterins.35 To evaluate the enzymatic synthetic pathway for BH4 synthesis involved in the insulin-induced enhanced NO production in TM cells, several inhibitors and stimulators of BH4 synthesis were tested in this study. Insulin's enhancing action on the NO production did not affected by dexamethsone, a known inhibitor of de novo synthetic pathway for BH4 synthesis. This finding suggests insulin could overwhelm the inhibitory action of dexamethasone on the NO production. However insulin's enhancing action on the NO production was markedly abolished by diaminopyrimidinone, another known inhibitor of de novo synthetic pathway for BH4 synthesis. Sepiapterin, acts as an inhibitor of de novo synthetic pathway for BH4 synthesis if not stimulated, also abolished insulin's enhancing action on the NO production.
Recently, several studies have shown that ascorbic acid, a stimulator of de novo synthetic pathway for BH4 synthesis, potentiates endothelial NO synthesis in a dose-time-dependent fashion36 and may protect NO from inactivation via chemical stabilization of BH4 in endothelial cells.37,38 Although ascorbic acid increased NO synthesis in this study, it abolished insulin's enhancing action on the NO production. Furthermore methotrexate, an inhibitor of salvage pathway for BH4 synthesis, markedly inhibited NO production despite of insulin's enhancing action on the NO production. These findings suggest that insulin enhances NO production in cultured human TM cells via de novo pathway of BH4 synthesis and ovewhelms the inhibitory action of glucocorticoid on the BH4-dependent endothelial function.
Preservation of the bioavailability of NO should be considered significant in the TM since NO is known to a cellular mediator that can modulate vascular tone as well as aqueous humor dynamic in the eye.39 Beside from its role in the regulation of trabecular outflow, another possible roles of NO also could be considered. The tissues facing the anterior chamber of the eye are exposed constantly to aqueous containing reactive oxygen products generated by light-catalyzed reactions. Such radicals may cause increased oxidation in tissues of the anterior eye segment and finally lead to oxidative damage. Oxidative stress that exceeds the capacity of the TM for detoxification could result in damage of TM cells and subsequent alteration of aqueous outflow resistance. Oxidative stress induces unavailability of BH4 and causes NOS to generate reactive oxygen substances. This will result both in NO depletion, and in peroxynitrite-dependent oxidation of BH4. Probably the resultant oxidative stress causes TM cell damage. Previous studies demonstrated decreased antioxidant potential of aqueous humor40,41 and increased oxidative DNA damage in the glaucomatous eyes.42,43 Chronic oxidative stress-induced TM cell damage has been suggested as an another possible pathogenetic mechanism of glaucoma recently.44
Recent report demonstrated the increased cellular senescence in the outflow pathway of glaucomatous eyes.45,46 Chronic oxidative stress, which resulted from NO dysregulation, also can provoke cellular senescence including TM cells.47,48 High glucose is known to cause cellular senescence and NO can delay cellular senescence caused by chronic oxidative stress.49,50 Thus the possible significance of preserving the bioavailability of NO in TM includes not only maintain trabecular outflow but also decrease oxidative stress, hence delay cellular senescence. Additional studies should focus on determining the most effective therapeutic approach for preventing oxidative stress and reducing the development and/or progression of glaucomatous optic neuropathy.
In summary, NO could decrease chronic oxidative stress of TM and delay aging of TM cells. This study suggests that insulin might play for a role in the maintenance of TM function by enhancing NO production via de novo pathway of BH4 synthesis. Further studies will be required to ascertain such roles of NO in TM.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print