Achondroplasia is the most common hereditary disorder that causes a dwarphism of prevalence 1/15,000 to 1/40,000, and this disorder is autosomal dominant [1]. Achondroplasia develops as a result of dysplasia of enchondral formation due to the mutation of fibroblast growth factor receptor 3 (FGFR3). The disorder is clinically characterized by such findings as low stature, an embossed frontal bone, craniofacial deformity, and vertebral malformation [2,3].
Ophthalmologic clinical characteristics include telecanthus, strabismus, abnormal anterior angle, Duane retraction syndrome, and the cone-rod retinal dystrophy [4-6].
We experienced a case of chorioretinal coloboma which was concurrently present in a child with achondroplasia. Here, we report our case and review the literature.
Case Report
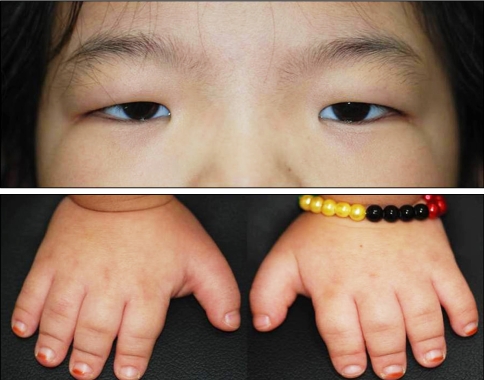
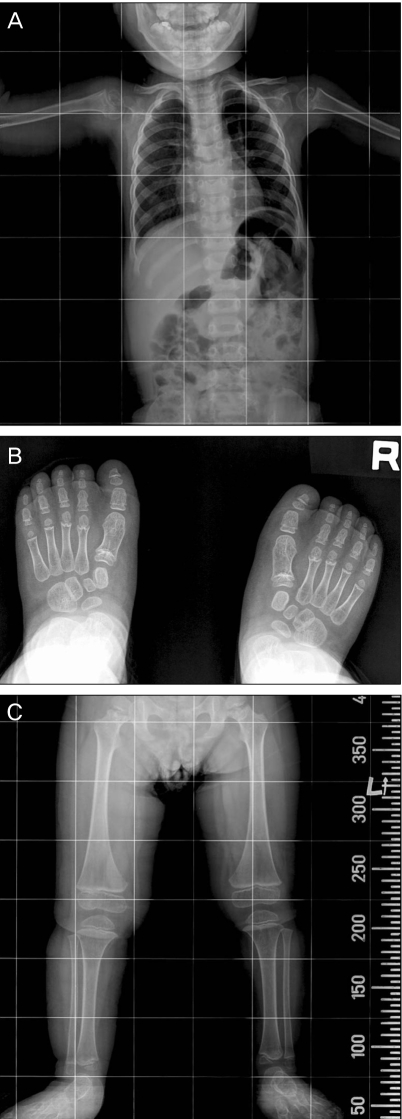
An 8-year-old girl visited us with a chief complaint of low visual acuity in both eyes. She was diagnosed with a developmental delay, known as achondroplasia, seven months after birth. She was born at term by vaginal delivery with normal birth weight (4.2 kg). There was no family history of hereditary ocular or systemic disease, and no history of drug intake by her mother. Grossly, the patient featured telecanthus and low nasal dorsum, and also had short, thick fingers (Fig. 1). Upon radiography, the intervertebral distance between the lumbar spine was narrowed, and the bones in the upper and lower extremities were typically short and thick (Fig. 2). There were no findings that are suggestive of mental retardation or hearing difficulty, and there were no specific findings upon brain magnetic resonance imaging and brain computed tomography.
At the time of initial visit, the visual acuity was 0.3 and intraocular pressure was 12 mmHg in both eyes. She demonstrated good eyeball movement and was orthophoric. Upon slit lamp examination, the notable findings regarding the anterior segment were not observed. Upon gonioscopic examination, both eyes had opened angles. The light reflex was normal in both eyes, and there was no relative afferent pupillary defect. Cycloplegic refraction revealed a refractive error of +3.00 diopter (D) in the right eye and +2.50 D in the left eye. Upon fundoscopy, the cup to disc ratio was increased to 0.8 in the right eye and 0.7 in the left eye, and bo th optic discs were colobomatous. In optical coherence tomography, both superior retinal nerve fiber layer showed some defects (Fig. 3). But visual field test could not be checked due to poor cooperation.
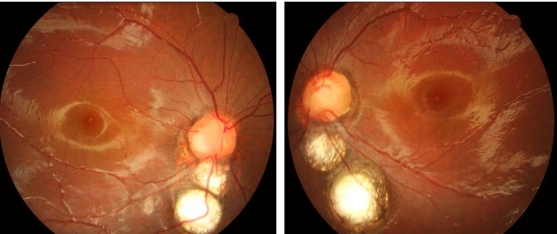
Symmetrically to both eyes, there were two optic disc-sized chorioretinal coloboma below the optic disc (Fig. 4). Upon fluorescein angiography, there were no notable findings other than hypoflurescent in the areas of chorioretinal coloboma (Fig. 5). However, when we tried electroretinography for evaluation of retinal dystrophy, she failed the test.
Thereafter, there were no changes in the fundoscopic findings in both eyes over 12 months. The best corrected visual acuity was 0.6 in both eyes. This patient was followed at the outpatient clinic.
Discussion
Achondroplasia is an autosomal dominant trait that occurs as a result of the mutation of genes encoding fibroblast growth FGFR3. Rosenthal et al. [4] described ophthalmologic characteristics in patients with achondroplasia. According to these authors, telecanthus was found in 26 (50%) of 52 patients and V type exotropia and bilateral inferior oblique muscle overaction were seen in 10 (20%) patients. Five patients presented with the tortousity of retinal blood vessels. In 26 of 46 patients, there were angle abnormalities, such as the definite presence of the iris process, and an incomplete sequestration and abnormal tissue at the anterior angle. Guirgis et al. [6] reported that Duane retraction syndrome and cone-rod retinal dystrophy were concurrently present in children with achondroplasia. Garg et al. [7] reported cases in which the fundus albipunctatus was identified in pediatric patients with achondroplasia.
Chorioretinal coloboma developed due to an in complete closure of the optic fissure during 6 to 7 weeks in the embryonic period. Also noted was the concurrent malformation of other organs [8]. Maumenee and Mitchell [9] proposed the hypothesis that this disease entity occurs due to the genetic defects of a single gene. These authors noted that it occurs in the early embryonic stage and features a high incidence of systemic and central organ malformation. Daufenbach et al. [10] reported retinal and choroidal detachment (8 eyes), microphthalmos (13 eyes), microcornea (6 eyes), 18 cases (38%) of systemic malformations, such as CHARGE syndrome, developmental delay, facial deformity, cleft palate, and transposition of the great vessel in 48 patients (86 eyes) with chorioretinal coloboma. According to these authors, due to the difficulty of the treatment of chorioretinal detachment, which can be concurrent with chorioretinal coloboma, the early detection and prophylactic management would be important. Because children are less likely than adults to be aware of and to report monocular visual loss, unilateral and even bilateral retinal or choroidal detachments in an already visually impaired child may remain undiagnosed for a long period of time. This diagnostic delay would make subsequent retinal detachment surgery less likely to be successful [10].
Herein, we report the correlation between achondroplasia and chorioretinal coloboma, which may be higher in the embryonic stage. It is, therefore, assumed that early and regular ophthalmologic examination would be essential to pediatric patients with achondroplasia.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print