Brain Imaging Studies in Leber's Congenital Amaurosis: New Radiologic Findings Associated with the Complex Trait
Article information
Abstract
Purpose
To report the incidence and new findings of abnormal brain imaging studies associated with patients initially diagnosed with Leber's congenital amaurosis (LCA) without definite systemic abnormalities and to determine the need for brain imaging studies in these patients.
Methods
A retrospective review of medical records was performed in 83 patients initially diagnosed as LCA and without definite systemic abnormalities before the age of 6 months in 2 tertiary referral centers. Brain magnetic resonance imaging was performed in 31 of 83 patients (37.3%).
Results
Six of 31 patients (19%) had radiologically documented brain abnormalities. Two patients had cerebellar vermis hypoplasia, 1 patient showed an absence of septum pellucidum, 2 subjects showed mild external hydrocephalus, and 1 patient was found to have a small cerebellum.
Conclusions
Approximately one fifth of the LCA patients in whom brain imaging was performed were associated with brain abnormalities, including the absence of septum pellucidum, which has not been documented in the literature. Brain imaging is mandatory in patients primarily diagnosed with LCA, even without definite neurologic or systemic abnormalities.
Leber's congenital amaurosis (LCA) is a hereditary congenital blindness usually presenting before 6 months of age, consisting of severe visual impairment, roving eye movements or nystagmus, extinguished or severely reduced scotopic and photopic electroretinogram (ERG), abnormal or variable visual evoked potentials and variable fundus examination results [1,2].
LCA is frequently associated with ocular or systemic abnormalities, including oculomotor apraxia, deafness, renal anomalies, skin or skeletal abnormalities, microcephaly, and developmental delay or mental retardation [2-4]. A reappraisal of previously diagnosed LCA patients revealed that 40% had an ocular or systemic disorder other than LCA, such as congenital stationary night blindness, achromatopsia, early-onset retinitis pigmentosa, Joubert syndrome, Zellweger syndrome, or infantile Refsum's disease [4]. Variable degrees of neurologic abnormalities are reported in up to 78% of LCA patients [3-8]. Therefore, a thorough systemic work-up is necessary in a patient diagnosed with LCA.
LCA is primarily a degenerative retinal disease, and magnetic resonance imaging (MRI) findings from a previous study demonstrated that blindness in LCA patients is not related to abnormality of the primary visual pathway, as the primary visual pathway signals were found to be normal in all the subjects and infants as young as 2 to 3 months, confirming that myelinization of this pathway had occurred at the correct age [3]. Therefore, brain imaging studies are not always recommended unless signs of neurologic deficiencies are present. However, as LCA patients usually present before 6 months of age, neurologic abnormalities may not be clearly defined in a blind subject when the developmental stages are mostly immature [2,9].
The authors of the present study reviewed the medical records of 83 patients initially diagnosed as LCA before the age of 6 months without definite neurologic or systemic abnormalities in 2 tertiary referral centers. The incidence of abnormal brain imaging in the patients was investigated to determine the need for imaging studies. Additionally, the clinical characteristics of LCA patients associated with specific radiological brain abnormalities, such as absent septum pellucidum or cerebellar vermis agenesis are described. Based on a comprehensive Medline literature search, the present study is the first report of absent septum pellucidum associated with LCA.
Materials and Methods
The medical records of 83 patients initially diagnosed as LCA before 6 months of age, in the department of ophthalmology of 2 tertiary referral centers, were evaluated. All patients fulfilled the diagnostic criteria currently proposed by De Laey [1] and a search for metabolic or degenerative central nervous system disorders was performed in all patients. Patients initially diagnosed with alleged neurologic or systemic abnormalities, or patients whom revealed any other hereditary retinal disease during follow-up were excluded. Brain MRI was performed in 31 patients who consented to the examination (37.3%). The described research adhered to the tenets of the Declaration of Helsinki and approval to conduct this study was obtained from the Institutional Review Board.
Results
Incidence of radiological brain abnormalities
Out of 31 patients, 6 patients (19.4%) had radiologically documented brain abnormalities. Two patients had cerebellar vermis hypoplasia with the classic "molar tooth" sign. One patient had absent septum pellucidum. Other alterations on MRI with unknown clinical significance were found in 3 subjects, which included a small cerebellum and 2 subjects with mild external hydrocephalus. The following cases depict the clinical characteristics of LCA patients associated with specific radiological brain abnormalities, including absent septum pellucidum and cerebellar vermis hypoplasia.
Case 1. Absent septum pellucidum
A 4-month-old girl was referred for poor eye contact. She was delivered preterm at 35 weeks of gestation after an uneventful pregnancy. She was born from healthy non-consanguineous parents and had an unremarkable family history. The patient did not fixate with the eyes or follow objects. Esotropia and nystagmus were present. Slit lamp examination and fundus examination showed no significant findings. Cycloplegic refractive errors were -1.50 D sph = -3.00 D cyl × 180° OD, -3.50 D sph = -3.00 D cyl × 180° OS. ERG was extinguished in both eyes and she was diagnosed as LCA. Systemic evaluation for metabolic abnormalities, as well as of the kidney, liver, and skin were unremarkable. Genetic analysis of mutations in CRX, CRB1, GUCY2D, AIPL1, RDH12, RPE65, LRAT, RPGRIP1, and TULP1 were unremarkable. Development was within the normal limit and neurologic evaluation was unremarkable at the time of diagnosis. However, brain MRI showed a prominent enlargement in the subarachnoid space of both frontal convexities, and the septum pellucidum was absent. No focal lesions were present in the brain and the size of the corpus callosum and white matter myelination was normal. At the age of 15 months, a follow-up brain MRI was taken, and external hydrocephalus with absent septum pellucidum was stationary (Fig. 1). A diagnosis of septo-optic dysplasia was considered, however, normal appearance of the optic nerve and normal pituitary function left the diagnosis doubtful. Strabismus surgery was performed for an esotropia of 40 prism diopters (PD). At the age of 3 years, the patient could still not fixate with the eyes and nystagmus was present, while ocular alignment was orthotropic. Cognitive and motor development was moderately delayed approximately 10 months, yet was slowly improving. The patient's medical history was unremarkable for myoclonic seizure.
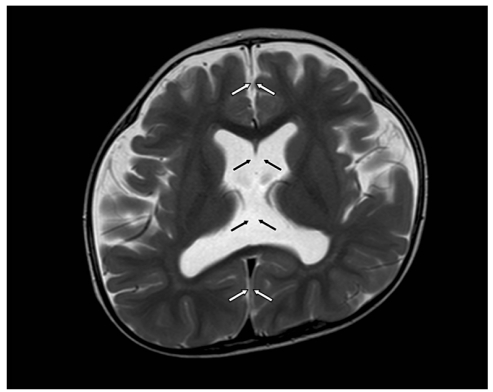
The T2 weighted axial magnetic resonance imaging of a Leber's congenital amaurosis patient (case 1) shows the absence of the septum pellucidum, which should normally be present in the area indicated between the black arrows. The hemispheric fissure is intact and indicated by the white arrows.
Case 2. Cerebellar vermis hypoplasia
A 4-month-old boy was referred for poor eye contact. He was born after an uneventful pregnancy, and his medical history was unremarkable in the perinatal period. The patient could not fixate with the eyes, and nystagmus was present. Exotropia of 15 PD was observed. Slit lamp examination and fundus examination showed no significant findings. Cycloplegic refractive errors were + 8.0 D sph OU. ERG was extinguished in both eyes, and the patient was diagnosed with LCA. Development was within normal limits and neurologic evaluation was unremarkable at the initial presentation. However, brain MRI revealed hypoplasia of the cerebellar vermis with enlarged 4th ventricles. Systemic evaluation for metabolic abnormalities, as well as of the kidney, liver, and skin were unremarkable. Genetic analysis of mutations in CRX, CRB1, GUCY2D, AIPL1, RDH12, RPE65, LRAT, RPGRIP1, and TULP1 were negative. At 8 years of age, development was within normal limit. His visual acuity was at the level of light perception in both eyes, and nystagmus had diminished; however, ERG was still extinguished.
Discussion
Pathologic findings in brain MRI of LCA subjects with systemic or neurologic abnormalities are documented in up to 33% of patients [3,5-7]. While most of the findings are mild and nonspecific alterations on MRI, such as slight reduction of the dimensions of the chiasm and/or optic nerve or white matter alterations, 2 to 10% of patients reveal pathologic findings of hypoplasia of the cerebellar vermis [3,5]. Without consideration of mild nonspecific alterations in brain MRI, the present study showed similar results to former reports, as 7% of primary LCA subjects were found to have pathologic brain imaging; half of the patients showing cerebellar vermis hypoplasia and the other patients revealing mild external hydrocephalus or absence of septum pellucidum, of which the latter is a novel finding in the present study. Notably, the perinatal history or neurologic examinations of the patients were unremarkable at the time of presentation before 6 months of age. Moreover, as the brain MRI was performed in only 37% of all patients who consented to the examination, the true incidence of abnormal brain structures may be larger. Waugh et al. [10] found that a significant amount of brain pathology occurs in children with congenital disorders of the peripheral visual system and the number of lesions varies directly with the degree of visual impairment and both correlate with developmental outcome. Therefore, brain imaging studies in patients initially diagnosed as LCA subjects should be mandatory, even if systemic or neurologic abnormalities are not obvious.
Case 1 and 2 showed MRI findings known to be a part of syndromic associations, namely septo-optic dysplasia with absent septum pellucidum and Joubert syndrome with cerebellar vermis hypoplasia [11,12]. However, the cases differed in many aspects from the typical syndromic characteristic of septo-optic dysplasia or Joubert syndrome [11,12]. In case 1, absence of the septum pellucidum without any other radiologic abnormalities lead to a differential diagnosis of septo-optic dysplasia [11]. The clinical picture of absent septum pellucidum and congenital nystagmus overlapped with the clinical and radiological features of septo-optic dysplasia [13]. On the other hand, the present case differed from typical septo-optic dysplasia in a number of specific aspects, i.e., the absence of hypoplasia of the optic disc, the absence of pituitary abnormalities and the absence of typical neurological impairment (e.g., myoclonic seizure was not present in our LCA subject). The absence of septum pellucidum may be an isolated finding and, although a characteristic sign of septo-optic dysplasia, it is not pathognomonic [14]. Congenital absence of the septum pellucidum is not associated with significant intellectual, behavioral, or neurological deficits in the majority of patients with septo-optic dysplasia [15]. Thus, the clinical significance of an absent septum pellucidum in case 1 with mild developmental delay is unknown. Case 1 may be considered as a type of complex LCA associated with the absence of septum pellucidum and mild developmental delay.
In case 2, the "molar tooth" sign of cerebellar vermis hypoplasia was present. Cerebellar vermis hypoplasia is found in up to 20% of brain imaging findings in LCA subjects [3,7,12]. The "molar tooth" sign is now recognized as present not only in Joubert syndrome but also in various cerebellooculo-renal syndromes (Arima, COACH, and Senior-Loken) [16,17]. The Joubert syndrome and related disorders are frequently associated with an LCA-like ocular phenotype [18]. Therefore, looking for systemic involvement in LCA patients with cerebellar vermis hypoplasia, including the kidney, liver and skin, to allow the delineation of LCA subtypes, is crucial [3]. However, the two LCA patients in the present study with cerebellar vermis hypoplasia differed significantly from the typical characteristics of Joubert syndrome-related disorders in a number of specific aspects, i.e., the absence of episodes of apnea-tachypnea, the absence of oculomotor apraxia-like head thrust, less severe neurological involvement (e.g., minimal hypotonia), and the absence of systemic involvement [19]. Thus, the subjects (including case 2) could be considered as a type of complex LCA associated with cerebellar vermis hypoplasia or another entity of Joubert syndrome-related disorders with only the ocular LCA phenotype. In a previous report, mutations in CEP290 were found in LCA patients associated with mental retardation or neurodevelopmental degeneration in the absence of the other systemic features of Joubert syndrome [20]. These individuals may represent a genetic subtype of LCA or an undiagnosed systemic disorder which remains to be elucidated [3]. Unfortunately, the genetic analysis of the CEP290 mutation was not available in the present study. Further studies and genetic evaluation are required to define this heterogeneous disease.
In conclusion, LCA has complex phenotypic features, and an absence of septum pellucidum can be associated with LCA, which has not been documented in the literature. Neurologic abnormalities may not be definite in a blind subject, especially under 6 months of age. Thus, brain imaging is mandatory in patients primarily diagnosed with LCA in the absence of definite neurologic abnormalities.
Acknowledgements
This study was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (A050488).
Notes
No potential conflict of interest relevant to this article was reported.