Vogt-Koyanagi-Harada (VKH) disease is a multisystemic autoimmune disease, mediated by T cells directed against melanocytes [1]. VKH disease causes granulomatous inflammation in the central nervous system, auditory system, integumentary system, and the eyes. The prodromal symptoms include neurologic and auditory symptoms. The initial acute phase then follows with iconic bilateral serous retinal detachment (SRD) with fulminant granulomatous choroiditis with or without inflammation of the anterior chamber and/or vitreous [2]. Use of high-dose corticosteroids is the mainstay treatment during this stage of the disease [3]. The eyes become quiescent without clinically conspicuous inflammation but transit to the convalescence stage with adequate treatment. Some eyes suffer a recurrence of uveitis in the form of granulomatous uveitis, which can develop into chronic recurrent VKH disease with a poorer prognosis. The proportion of patients with chronic evolution of initial-onset VKH disease after systemic corticosteroid monotherapy is about two-thirds in the international population and one-fourth in the Japanese population, suggesting less severe disease in Japan [4]. The rate is not known for Korean patients.
Recurrent VKH is more prominent in anterior segments, with more prevalent mutton-fat keratic precipitates, cells and flare in the anterior chamber, posterior synechiae, and iris nodules, and less prominent SRD [5]. Recurrent anterior uveitis is associated with higher complication rates of cataracts, glaucoma, and neovascular membranes, for which stronger and prolonged systemic immunosuppression and additional treatments are needed [5-8]. More prolonged and stronger immunosuppression at the time of the initial acute-onset VKH disease may help to reduce the rate of chronic recurrent evolution of the disease when risk factors for recurrent anterior uveitis are considered.
Here, we included patients with initial acute-onset VKH disease at two university hospitals and focused on the recurrence of VKH disease in the form of anterior granulomatous uveitis, after uveitis had disappeared for at least 3 months. We calculated the estimated incidence of recurrent anterior uveitis in patients with VKH disease by survival analysis. The risk factors were identified from multivariate Cox regression analysis. We included patientsŌĆÖ demographic data, underlying conditions, ocular characteristics, and treatment methods in the analysis to calculate hazard ratios (HRs).
Materials and Methods
This study was approved by the Institutional Review Board of Pusan National University Hospital (No. 2204-014-114) and Pusan National University (No. 2023-02-015). The requirement for informed consent was waived due to the retrospective nature of the study. The authors adhered to the tenets of the Declaration of Helsinki throughout the study.
Patients who were diagnosed and treated for initial acute-onset VKH disease during 2003-2022 at Pusan National University Hospital, and during 2010-2022 at Inje University Haeundae Paik Hospital, were retrospectively recruited. Past medical and surgical histories were collected from the medical records. The results of systemic evaluation for possible causes of uveitis and comprehensive ophthalmologic examinations were reviewed to rule out other possible causes for the uveitis. The diagnosis of VKH disease followed the revised criteria for VKH disease by the International Committee on VKH Disease, published in 2001 [2]. Patients who were diagnosed as VKH disease in another hospital and had already received systemic corticosteroid treatment were excluded. Patients who had ocular symptoms more than 6 weeks before the first visit were also excluded. Patients who had been treated for a previous episode of VKH disease and/or showed clinical manifestations of chronic recurrent granulomatous anterior uveitis were also excluded. Patients who maintained at least 3 months of no conspicuous uveitis and complete resolution of SRD, with or without treatment, were included in the analysis.
The demographic characteristics of the patients (age and sex), underlying diseases (diabetes and hypertension), presence of prodromal symptoms (neurologic or auditory), and duration of visual symptoms were retrospectively collected from the medical records. Initial best-corrected visual acuity (BCVA), slit-lamp findings (anterior chamber cells and flare by Standardization of Uveitis Nomenclature (SUN) Working Group grading scheme [9], keratic precipitates, and focal posterior synechia), fundus findings (presence of SRD and optic nerve hyperemia/papillitis), fluorescein angiography (FA) findings (disc hyperfluorescence), and optical coherence tomography (OCT) measurements (height of SRD) were documented. The initial day of high-dose systemic steroid therapy and data of the systemic treatment regimen and duration were also included. Dates of the disappearance of SRD, recurrence of SRD during steroid tapering, and recurrent anterior uveitis were investigated.
Recurrent anterior uveitis was defined as the first occurrence of the granulomatous anterior uveitis with anterior chamber cells and flare of 2+ or more by SUN Working Group grading scheme [9], after the disappearance of conspicuous uveitis and SRD (on OCT) for at least 3 months, regardless of systemic or local treatment. The event was defined as recurrent anterior uveitis. The time for survival analysis was defined as the period from the diagnosis to the event when anterior uveitis had occurred, and from the diagnosis to the last visit when no event had occurred. Recurrence of SRD (posterior choroiditis) during tapering of the systemic steroid was defined as rebound SRD during steroid tapering. Papillitis was defined as disc swelling and hyperemia on the fundus examination and the late leakage and stain of the disc on FA.
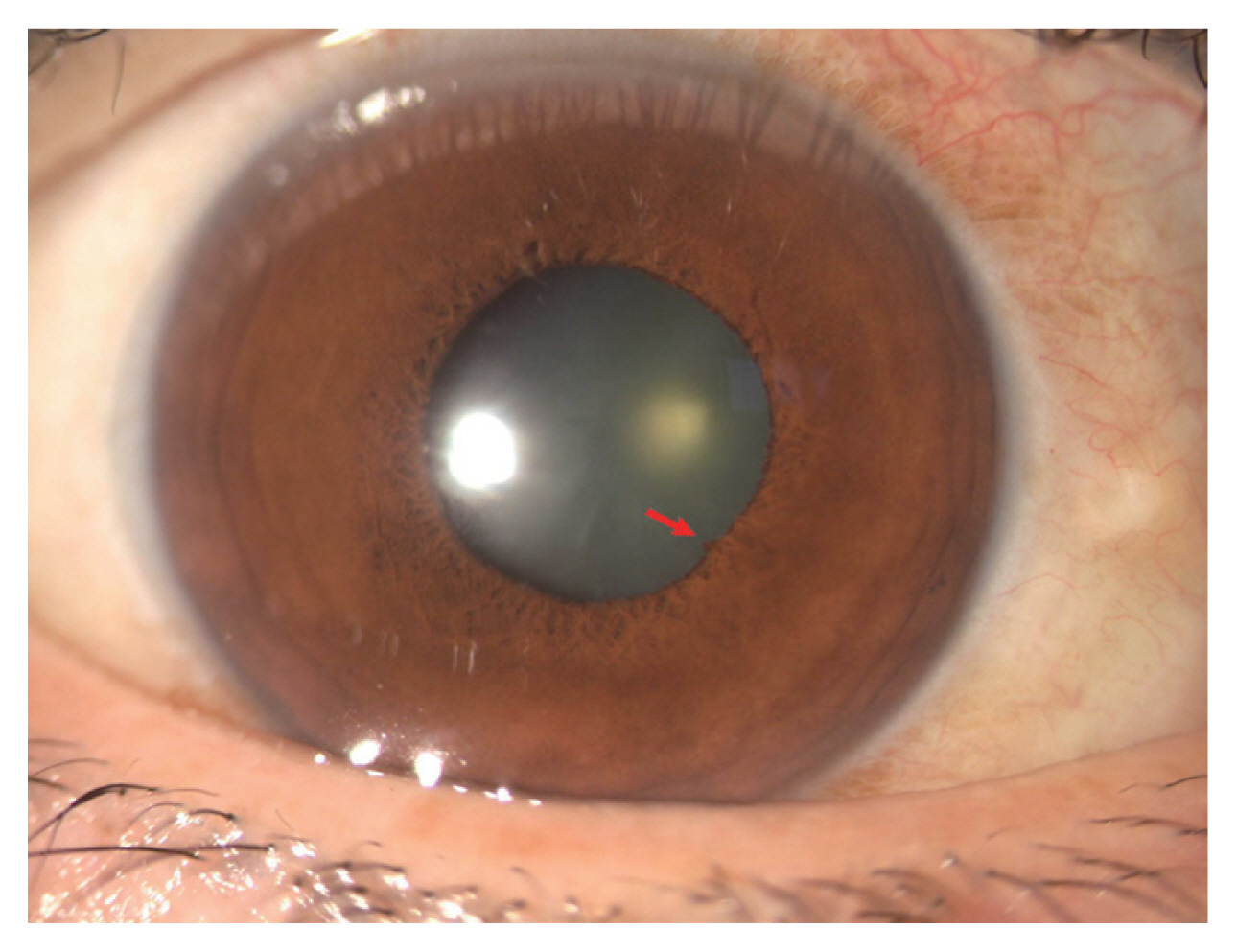
SRD height was defined as the farthest distance from the retinal pigment epithelium to the outer margin of the sensory retina within six macular radial scans of Stratus OCT (Carl Zeiss Meditec), within 6 ├Ś 6-mm macular cube scan (centered in the fovea) of Cirrus OCT (Carl Zeiss Meditec), Spectralis OCT (Heidelberg Engineering), and Triton OCT (Topcon Medical Systems). The peak point of SRD was occasionally not over the fovea, so the highest SRD was measured within the 6 ├Ś 6-mm area. SRD height was entered into the analyses as a nonparametric variable (whether or not it exceeded 700 ╬╝m). The presence of anterior chamber cells and keratic precipitates was determined from the medical records. Focal posterior synechiae was defined as a very focal posterior synechia at the diagnosis without a history or signs of anterior uveitis, which were observed concurrently during acute-stage VKH disease, with a few keratic precipitates and anterior chamber cells of 2+ or less and fulminant posterior choroiditis with SRD (Fig. 1). The focal posterior synechiae disappeared during systemic corticosteroid therapy. VKH disease always involves both eyes, thereby all nonparametric variables were considered positive, even when they involved only one eye. Recurrence of anterior uveitis always involved both eyes, although the severity of the uveitis between the eyes varied occasionally. The time to initiate high-dose corticosteroid therapy was defined as the period between the onset of visual symptoms to the day therapy was initiated. Mean BCVA was defined as the average of BCVA of the two eyes in logarithm of the minimum angle of resolution (log-MAR).
The subfoveal choroidal thickness could not be measured before the equipment of enhanced depth imaging OCT (EDI-OCT) in 2014 at both hospitals; thus, the information of choroidal thickness could not be used in analyses. Integumentary complications and sunset-glow fundus are signs of chronic stages and were included in the diagnostic criteria for complete VKH disease [2]. However, they are not the signs of initial acute VKH disease; thus, they were not included in the analysis as presumed risk factors for recurrent a nterior uveitis. Complication rates of cataracts and increased intraocular pressure (IIOP) during follow-up were investigated. Diagnosis of complete, incomplete, and probable VKH diseases was made retrospectively for incidence.
The Kolmogorov-Smirnov test was performed to test the normality of the distributions of continuous variables. The Kaplan-Meier log-rank test was performed for univariate analyses of presumed risk factors. Parameters with p < 0.20 in univariate analyses were entered into multivariate Cox regression analysis to calculate the HRs. Age and mean initial BCVA were entered into the analysis as continuous variables. The independent t-test was used to compare the final BCVA between the patients who had recurrent anterior uveitis and those who did not. IBM SPSS ver. 22.0 (IBM Corp) was used for statistical analyses; p < 0.05 were considered significant.
Results
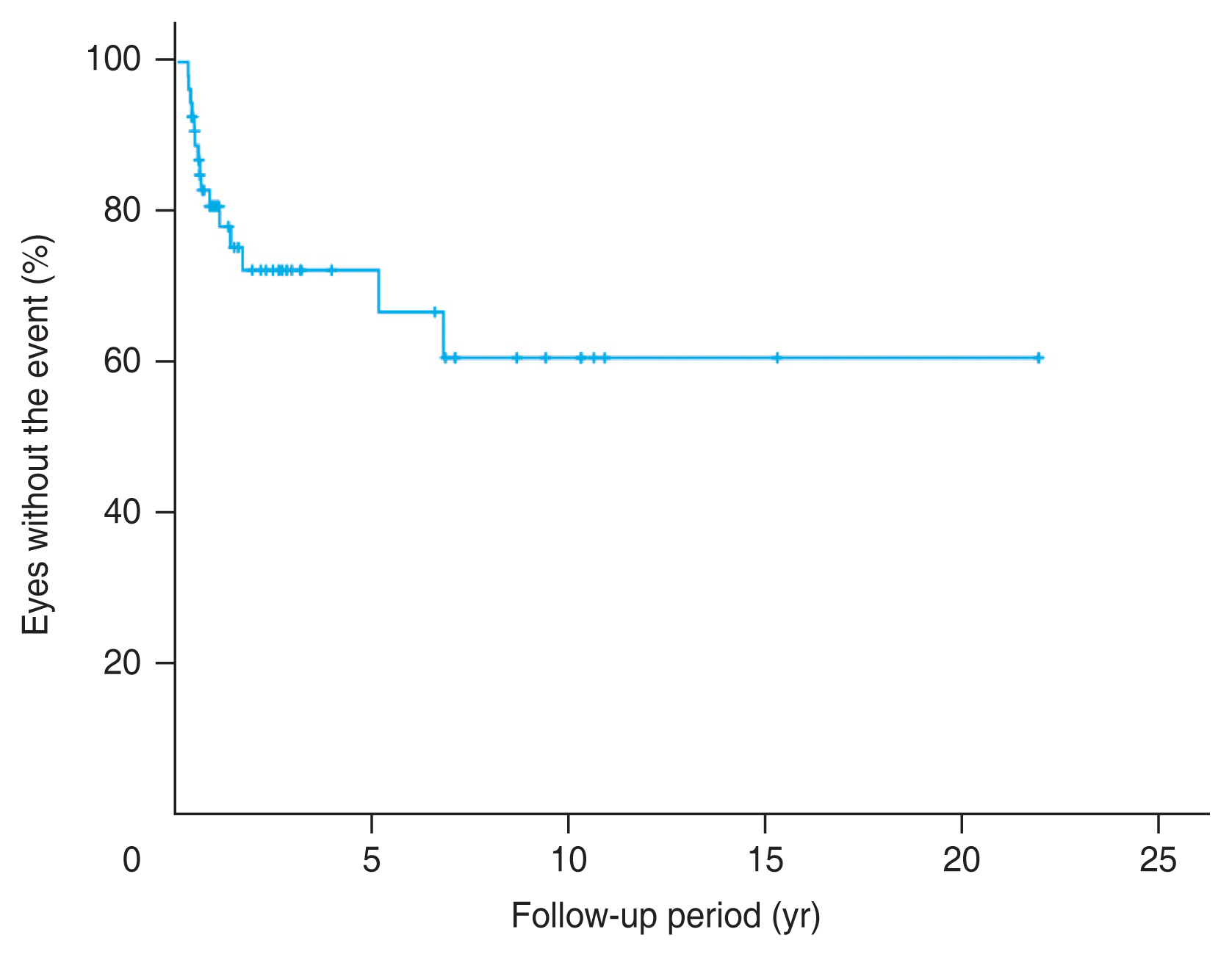
A total of 55 patients were included in the study. Of which, 15 patients (27.3%) developed recurrence of VKH disease in anterior segments during the mean ┬▒ standard deviation [SD] follow-up of 4.5 ┬▒ 7.0 years, with a range of 5 months to 22 years. The estimated incidence of recurrent anterior uveitis from the survival analysis was 39.3% at 10 years (Fig. 2). The event occurred from 3.8 months to 5.2 years (mean ┬▒ SD, 1.4 ┬▒ 1.9 years) after the diagnosis of initial acute VKH disease.
The mean ┬▒ SD age of the patients was 44.3 ┬▒ 11.4 years (range, 16.6-67.1 years) and 32 patients (58.2%) were female. Prodromal symptoms such as headache, meningismus, vertigo, and dysacusis were present in 39 patients (70.9%). Patients suffered visual disturbances for 1 to 30 days (mean ┬▒ SD, 8.1 ┬▒ 7.5 days) before the initial visit, and the mean ┬▒ SD initial BCVA was 0.64 ┬▒ 0.51 in logMAR. Four patients (7.3%) had diabetes and 10 patients (18.2%) had hypertension (Table 1).
Forty-eight patients (87.3%) started intravenous high-dose steroid pulse therapy (methylprednisolone 1,000 mg/day for 3-5 days), three patients (5.5%) received high-dose oral corticosteroid 1 to 2mg/kg/day within 1 to 39 days since the visual symptoms developed. Twenty-seven patients started high-dose steroid therapy (intravenously in 25 patients and orally in two patients) more than 7 days after visual symptoms developed. Six patients (10.9%) received an adjuvant intravitreal triamcinolone acetonide (IVTA; 4 mg/0.1 mL) during tapering of the oral-systemic corticosteroid. Immunosuppressive agents (oral cyclosporin 100 mg twice a day) were administered with the oral-systemic steroid during the initial acute stage in four patients (7.3%). Keratic precipitates and focal posterior synechiae were observed in eight (14.5%) and six patients (10.9%), respectively. High SRD (>700 ╬╝m) was detected in 17 patients (30.9%), and all patients in the study had SRD involving the macular area. Optic disc hyperemia/papillitis was detected concurrently in 26 patients (47.3%).
Table 1 shows the results of the univariate log-rank test. A medical history of diabetes was associated with an increased risk for recurrent anterior uveitis, although only three patients had the event (p = 0.029). The presence of focal posterior synechiae at the diagnosis was associated with an increased risk in the univariate analysis (p < 0.001). Rebound SRD during steroid tapering was also relevant to a higher risk for recurrent anterior uveitis ( p = 0.025). Time to initiate high-dose steroid therapy more than 7 days after the development of visual symptoms was related to an increased risk for recurrent anterior uveitis (p = 0.019).
The HRs from the multivariate Cox regression analysis are shown in Table 2. The presence of focal posterior synechiae at the diagnosis increased the risk of recurrent anterior uveitis 6.97-fold compared to the absence of synechiae (95% confidence interval [CI], 2.20-22.11; p < 0.001). The calculated HR for use of systemic high-dose steroid therapy more than 7 days after the visual symptoms developed was 4.55 (95% CI, 1.27-16.40; p = 0.020).
A sunset-glow fundus and/or chorioretinal atrophic scars developed in 36 patients, and the integumentary signs were noted in 16 patients. Late complications of cataracts were present in 10 patients (18.2%), and 13 patients (23.6%) were treated for IIOP. Complete VKH disease was diagnosed in 37 patients (67.3%), incomplete disease in 13 patients (23.6%), and probable disease in five patients (9.1%). The mean ┬▒ SD BCVA at the final visit was 0.13 ┬▒ 0.17 in log-MAR for all patients. The mean BCVA of the patients who had recurrent anterior uveitis at the final visit (mean ┬▒ SD, 0.19 ┬▒ 1.19 in logMAR) was worse than that of patients who did not have the event (mean ┬▒ SD, 0.11 ┬▒ 0.16 in log-MAR), but was not statistically significant.
Discussion
This study showed that the actual incidence of recurrent anterior uveitis was 27.3% among Korean patients, which was similar to that of the Japanese population [4]. However, the estimated incidence from the survival analysis, which accounted for the loss of follow-up among study patients, was higher (39.3% at 10 years). The risk factors for recurrent anterior uveitis in the multivariate Cox regression were focal posterior synechiae at the initial presentation and delayed application of systemic high-dose steroid therapy.
Maruyama et al. [10] defined recurrent VKH disease as the need for additional treatment, such as immunosuppressants for recurrent inflammation, or other complications, such as SRD or choroidal neovascularization. The need for a steroid only therapy was regarded as nonrecurrent. They reported 22 patients (53.7%) with recurrence among the 41 patients who were treated with the same systemic steroid tapering protocol. Flare in the anterior chamber measured with a flare meter, choroidal thickness assessed by swept-source OCT, and the mean blur rate evaluated with laser speckle flowgraphy were compared between the recurrent and nonrecurrent groups. They found a lower BCVA, a higher initial flare number, and a lower response of mean blur rate to treatment in the recurrent group than in the nonrecurrent group. The strength of that study was that they used the same treatment protocol for all study subjects and used objective methods to quantify the parameters.
Chee et al. [11] included 29 patients and focused more on the prognosis of VKH disease. They defined resolved VKH disease as a state with no sunset-glow fundus, perilimbal vitiligo, peripapillary atrophy, or clinical activity; the rest were defined as a chronic disease. They reported that predominant swelling of the optic disc at 2 months was related to chronic or chronic recurrent disease, and lower central retinal thickness measured by OCT at 1 week and the persistently high central retinal thickness measured at 2 months were related to chronic recurrent disease. Another study showed that early pinpoint peripapillary hyperfluorescence on FA was a significant risk factor in multivariate analysis for chronic recurrent VKH disease [12].
The outcome measures, the analyzed parameters, and the definition of recurrent VKH disease vary among previous studies. The chronic recurrent phase is characterized by episodes of granulomatous anterior uveitis [13]. This phase is more refractory to treatment and is associated with a higher rate of complications; thus, this subset of VKH patients has poorer functional and anatomical outcomes [14]. Accordingly, we determined recurrent anterior uveitis as the event and primary outcome measure in this study. Recurrence of SRD during tapering of systemic corticosteroid is reported to have good visual prognosis [15]; rebound SRD during steroid tapering in this study was not a risk factor for recurrent anterior uveitis from the multivariate Cox regression analysis.
The major limitations of this study are that it is retrospective, and that only a small number of patients were included from the two hospitals over 10 to 20 years. The incidence of VKH disease has been estimated to be 6.5 per million in a nationwide survey in Japan [16]. Although VKH disease usually affects more pigmented races, such as Asian, Middle Eastern, Native American, and Hispanic population, with a predisposing genetic background [17,18], the overall incidence in Japan shows that VKH disease is not common. Additionally, choroidal thickness and blood flow data were not entered into the analysis because of a lack in data (choroidal thickness was measured in only 20 patients [36.4%]). Many studies have emphasized the utility of OCT and OCT angiography in recurrent VKH disease [19-22]. The predictive value of EDI-OCT for monitoring the recurrence of VKH disease has been emphasized, because an increase in choroidal thickness can be detected during and before the recurrence of VKH disease [19].
Acute-stage VKH disease manifests with bilateral SRD, but concurrent low-grade anterior inflammation can also occur [2]. The evidence of anterior uveitis, i.e., keratic precipitates and anterior chamber cells of 0.5+ to 2+ simultaneously present in the eyes with focal posterior synechiae, disappeared in response to systemic steroid therapy. Multivariate analysis considering the possible confounding effects among the parameters showed that the presence of focal posterior synechiae was a major risk factor for recurrent anterior uveitis (HR, 6.97; 95% CI, 2.20-22.11; p < 0.001). The presence of focal posterior synechiae with SRD at the initial phase of acute VKH disease may reflect the state of panuveitis. In other words, it may be assumed that a broader involvement of inflammation in uveal tissue at the diagnosis may lead to more frequent recurrence. However, because of the retrospective nature of this study, it is hard to confirm the consistency of the medical records regarding risk factors. Thus, the presence of focal posterior synechiae can be inconclusive as a risk factor, and further studies are warranted.
There is a consensus that early diagnosis and prompt and adequate immunosuppression halts the progression of the disease and prevents recurrence [23,24]. Use of immunosuppressants, such as azathioprine, cyclosporine, and mycophenolate mofetil, with a systemic corticosteroid, has been suggested to help in preventing recurrences [17,25-27]. Use of a high-dose systemic steroid (intravenously in 25 patients and orally in two patients) for more than 7 days after the development of visual symptoms increased the risk for recurrent anterior uveitis about 4.5-fold (HR, 4.55; 95% CI, 1.27-16.40; p = 0.020). Ono et al. [28] reported that oral combination therapy with prednisolone and cyclosporine was not inferior to intravenous corticosteroid pulse therapy. In this study, the use of immunosuppressive agents with an oral steroid did not affect the risk of recurrence, probably because the number of patients in whom the therapy was used was small. The use of IVTA during tapering of a systemic steroid for rebound SRD is effective [29] but did not affect the risk of recurrent anterior uveitis in this study.
Further prospective, multicenter studies or a population-based study are needed to identify risk factors for the recurrence of VKH disease and establish the proper treatment guidelines for patients at risk. Clinicians should always consider the possibility of recurrent anterior uveitis in these patients.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print