Dear Editor,
Ectopia lentis (EL) refers to displacement of a lens from its normal location, often when the fibrillin-rich microfibrils of the ciliary zonule are stretched. Marfan syndrome (MFS) is the most common systemic disease associated with EL [1]. MFS is an autosomal dominant connective tissue disorder with major involvement of ocular, skeletal, and cardiovascular systems [1]. The diagnosis of MFS is made on the basis of clinical and genetic criteria that are referred to as the Ghent criteria [1]. Mutations in the fibrillin-1 (FBN1) gene located on chromosome 15q12 are known to be causative for MFS [1,2,3]. The FBN1 mutations are associated with an increased risk of aortic dilatation and dissection, regardless of the clinical presentation [3]. We report a case of monozygotic twin boys presenting initially with juvenile-onset bilateral EL with disease-causing missense mutation in the FBN1 gene.
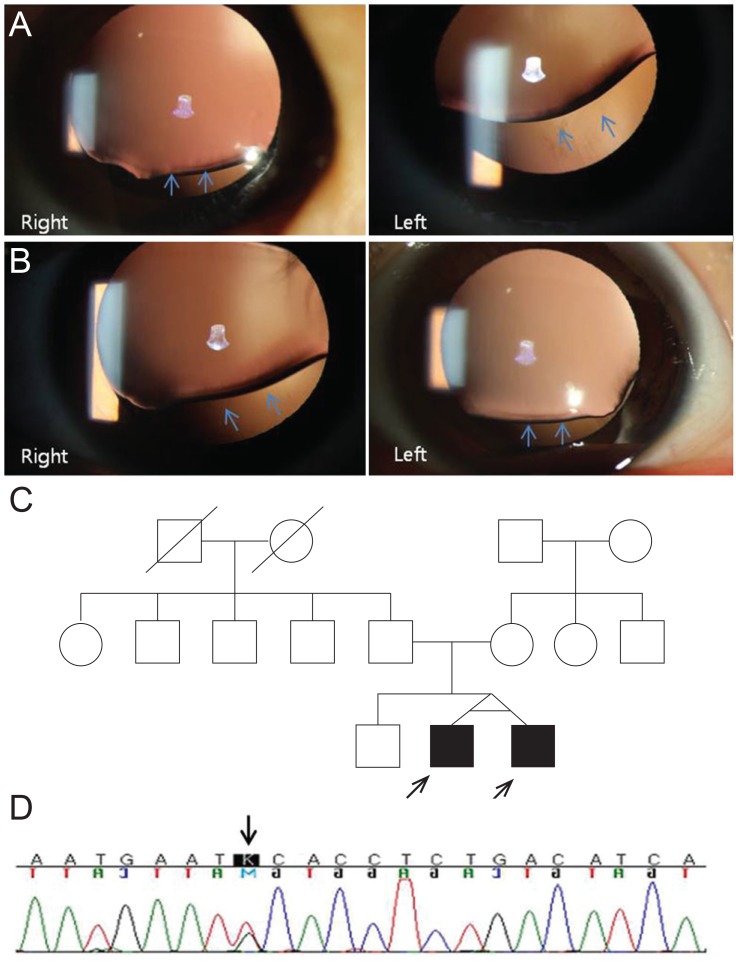
Five-year-old monozygotic twin boys were referred for evaluation of low vision in both eyes. They were born at 37 weeks gestation and weighed about 2.5 kg. They were the second birth of non-consanguineous parents. There was no previous family history of ocular, systemic, or chromosomal disorders. The first-born boy had best-corrected visual acuity (BCVA) of 20 / 50 in the right eye with a refraction of +2.00 Dsph = -4.50 Dcyl ├Ś 170┬░A and 20 / 120 in the left eye with a refraction of +1.00 Dsph = -7.75 Dcyl ├Ś 180┬░A. On slit lamp examination, the lenses of the right and left eyes were upwardly and superonasally dislocated, respectively, with the left eye having more severe displacement (Fig. 1A). The second boy's BCVA was 20 / 60 in the right eye with a refraction of +1.50 Dsph = -5.75 Dcyl ├Ś 10┬░A and 20 / 50 in the left eye with a refraction of +2.00 Dsph = -4.00 Dcyl ├Ś 10┬░A. The lenses were displaced superotemporally and upwardly in his right and left eyes, respectively (Fig. 1B). No other features of MFS were identified on initial examination. After obtaining informed consent from their parents, direct DNA sequencing of the FBN1 gene was performed. Direct sequencing of all exons of the FBN1 revealed a heterozygous missense mutation (c.7583G>T) in exon 62 which results in a change from cysteine to phenylalanine (Cys2528Phe) (Fig. 1C and 1D). This mutation was first reported in a French female patient with MFS but has not been previously reported in Korea [4]. It is predicted to be damaging as shown in Sorting Intolerant from Tolerant analysis (SIFT, http://sift.jcvi.org). This mutation was absent in their parents (Fig. 1C). Based on the molecular result, cardiac evaluation was recommended, and echocardiography was performed which revealed aortic root dilatation in both patients. They were prescribed a beta-blocker to prevent further enlargement of the aortic root. The amblyopia was successfully treated with spectacle correction of high astigmatism. After 1 year, the first-born boy had BCVA of 20 / 25 in the right eye and 20 / 60 in the left eye. The second boy's BCVA was 20 / 40 in the right eye and 20 / 32 in the left eye.
FBN1 encodes fibrillin-1, a cysteine-rich secreted glycoprotein that is a vital component of the extracellular matrix and the major structural element in the suspensory ligaments of the lens [1,2,3]. FBN1 mutations have been associated with a broad spectrum of phenotypes ranging from single connective tissue manifestations such as isolated EL, to lethal neonatal MFS [1,2,3]. It is interesting that a heterozygous missense mutation of c.7583G>T (Cys2528Phe) in exon 62, confirmed in our 5-year-old twin boys, was associated with juvenile-onset ocular and cardiovascular manifestations. Mutations in exons 59-65 were reported to be associated with major involvement of the cardiovascular system [5]. Our finding demonstrates the necessity of molecular analysis for FBN1 gene mutation in patients presenting with isolated EL. Molecular diagnosis enables proper and early identification of affected patients who need adequate follow-up and treatment for prevention of the life-threatening complications of MFS.
Upwardly d islocated lenses, found i n all of our cases, must be differentiated from lens coloboma, which represents incomplete lens formation due to failure of the fetal fissure to close completely. In our cases, the lens changes were not accompanied by other ocular colobomatous changes of the eyelid, iris, choroid, or optic disc. Anterior optical coherence tomography may be useful to visualize anterior segment structures. In this case, successful improvement of visual acuity with appropriate spectacles alone shows that appropriate optical correction should be tried first in children with EL. Careful and repeated measurements of phakic or aphakic refraction are necessary to achieve the best possible vision. Surgical procedures should be considered for the prevention of irreversible amblyopia when the dislocated lens edge bisects the pupillary axis or spectacle correction is impossible.
In conclusion, we described monozygotic twin boys presenting initially with juvenile-onset bilateral EL with disease-causing de-novo missense mutation in the FBN1 gene. In patients with isolated EL, molecular analysis for FBN1 gene should be performed for prompt diagnosis and early identification of cardiovascular complications. Furthermore, this study aids in expanding the genotype-phenotype association with respect to FBN1 mutation in MFS.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print