Retinal capillary hemangioblastoma (RCH) is a benign hamartoma of the retina and optic nerve. RCH occurs in a solitary form or as a syndromic form associated with von Hippel-Lindau (VHL) disease [1]. VHL is an autosomal dominant disorder caused by mutations in the VHL gene located on the short arm of chromosome 3, and its clinical manifestations include subtentorial hemangioblastoma, renal cancer, pheochromocytoma, and RCH [1]. RCH is the most frequent and the earliest manifestation of VHL disease [2]. RCH usually manifests as a solitary tumor; however, one-third of the patients with VHL have multiple retinal capillary hemangioblastomas, and up to one-half of patients have bilateral involvement [3].
RCH is a highly vascularized tumor that presents as a red or pinkish round mass with feeding arteries and engorged draining veins. Excessive exudation from vascular mass and glial proliferation can lead to exudative or tractional retinal detachment, vitreous hemorrhage, and macular preretinal fibrosis, resulting in visual deterioration [4-6]. Before these complications occur, small-sized RCH can be effectively treated with laser photocoagulation; therefore, early detection and proper treatment of RCH are essential for preserving visual function [7].
A recent study reported VHL as the underlying cause in 84% of patients with RCH [8]. Likewise, patients with VHL require lifetime screening and frequent surveillance for RCH and other tumors [9]. VHL genetic diagnosis is critical for surveillance, and molecular genetic testing has become the recommended standard procedure for suspected VHL [10]. Although new genetic testing technology has expanded the understanding of VHL, there are only a few studies on the mutation profile of the VHL gene and its clinical features in Korean patients with RCH [11-14]. This study aimed to investigate the clinical features of RCH patients and the genetic data of hereditary forms (VHL disease) in the Korean population.
Materials and Methods
This study was reviewed and approved by the Institutional Review Board of Seoul National University Hospital (No. B-2206-761-102). This study adhered to the tenets of the Declaration of Helsinki. Informed consent was waived due to the retrospective study design.
The medical records of 18 patients (23 eyes) with RCH treated between May 1, 2003 and December 31, 2021 at Seoul National University Bundang Hospital were reviewed retrospectively. The diagnosis was made based on typical fundoscopic and fluorescein angiographic findings.
All patients underwent complete ophthalmological examinations, including best-corrected visual acuity, intraocular pressure, slit-lamp, and fundus examinations. Fundus photography and fluorescein angiography were performed for all patients, and the following data were recorded: sex, age at diagnosis, tumor characteristics (number, location, and size), accompanying ocular disorder, and other organ involvement. Patients with one RCH were included solitary form [15]. The majority of RCHs are located in the peripheral retina, and can occur in the juxtapapillary region [1]. As for the location of tumor, it was classified into juxtapapillary region, the posterior pole, and the mid to far periphery. According to previous studies, VHL disease was diagnosed based on the presence of particular tumors, family history, and genetic testing [16-17]. If a patient has a confirmed family history of VHL disease, a diagnosis of VHL disease can be made by finding a single VHL tumor (e.g., hemangioblastoma, pheochromocytoma, renal cell carcinoma, pancreatic endocrine tumor, and endolymphatic sac tumor). Patients without a family history of VHL require the presence of two tumors (e.g., two hemangioblastomas or a hemangioblastoma and a visceral tumor). Clinical information was collected, including disease progression, complications, and treatment modality. Complications were counted by the number of eyes, not by the number of patients.
Blood sampling and DNA collection from patients and their families were performed. Multiplex ligation-dependent probe amplification (MLPA) and direct sequencing targeting whole regions of the VHL gene, including all exonintron junctions, were used for genetic testing. All types of mutational changes in the VHL gene, including single nucleotide variations, small insertions and/or deletions, and large deletions and duplications, were analyzed [11].
The Mann-Whitney U-test and Fisher exact test were used to compare the clinical features and demographics of sporadic RCH and VHL disease-related RCH. All statistical analyses were performed using R ver. 4.0.2. (R Foundation for Statistical Computing, Vienna, Austria). Statistical significance was set at p-value of <0.05.
Results
A total of 18 patients with RCH (10 male and eight female patients) were enrolled in this study. The mean age of the patients at diagnosis was 39 Âą 17 years. Twelve patients were diagnosed with VHL disease, while the remaining were considered sporadic cases of RCH (no family history of VHL, no systemic complications, and a negative or nonexistent genetic test result). Of the six patients who underwent genetic tests, pathogenic variants were identified in four patients. A pathogenic variant, c.208G>A (p.Glu70Lys), in the VHL gene was found in three patients with type 1 VHL disease, and a nonsense variant, c.264G>A (p.Trp88Ter), was identified in a patient with bilateral juxtapapillary RCH. Two patients whose genetic variation was not found in the MLPA test were classified as sporadic case. They also had no family history and no specific tumors. The clinical features are summarized in Table 1.
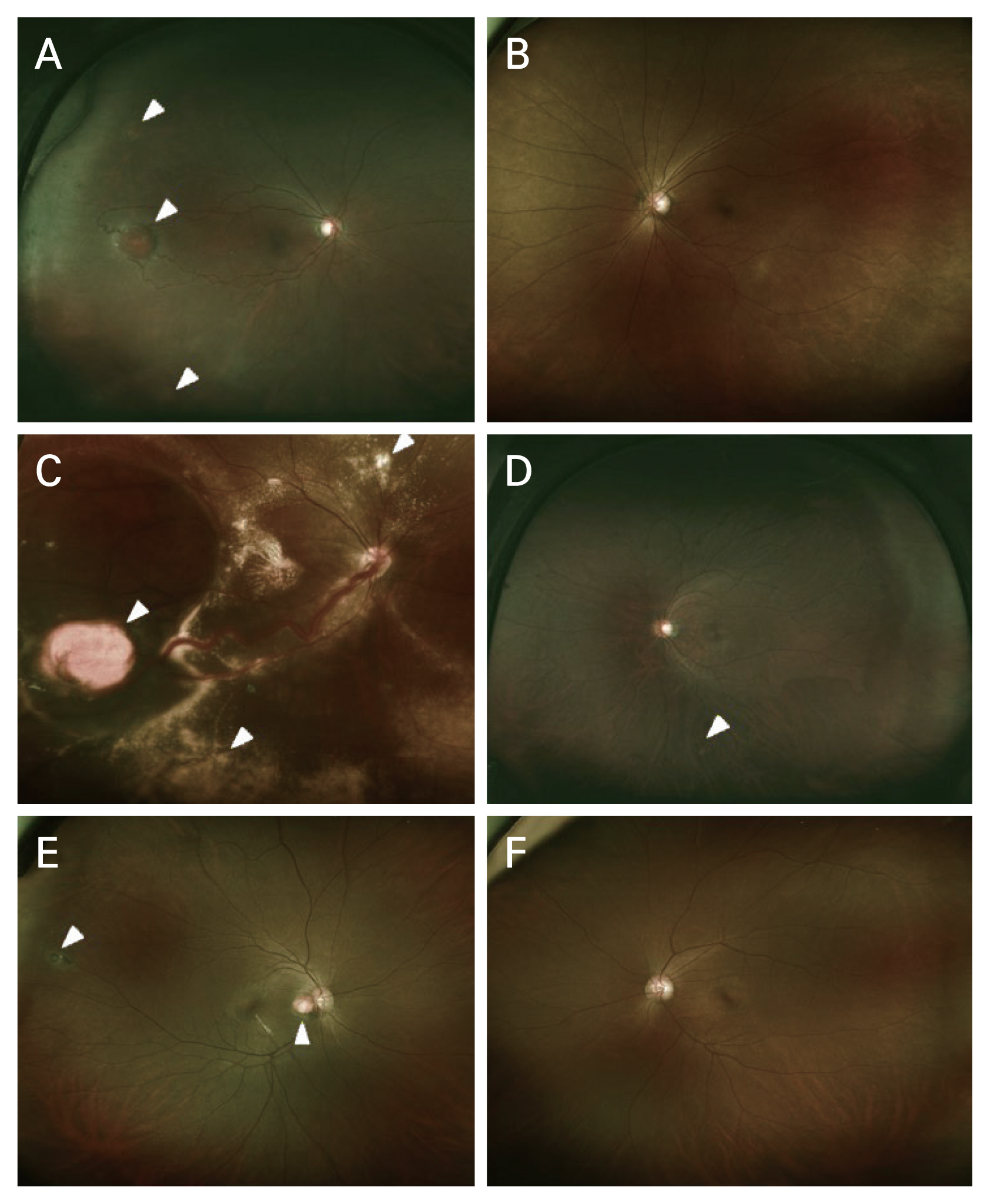
Five patients were diagnosed with bilateral RCH, and 13 patients had unilateral RCH. Average initial visual acuity in the eye with RCH was 0.32 Âą 0.46 logarithm of the minimum angle of resolution (logMAR; Snellen equivalent, 20 / 40), and at last follow-up was 0.80 Âą 0.93 logMAR (Snellen equivalent, 20 / 125). The most common complication with RCH was the epiretinal membrane (ERM), followed by subretinal fluid (Fig. 1A-1F). Retinal exudation or proliferative gliosis, which are the main causes of visual impairment in RCH, were observed in 11 eyes (50%) [3]. At the final follow-up, the mean visual acuity of the 11 eyes was 1.40 Âą 1.00 logMAR (Snellen equivalent, 20 / 500), of which eight eyes (73%) had legal blindness. Eleven eyes without complications or with stable ERMs showed stable visual acuity during the follow-up period. Eyes with exudative or tractional retinal detachment (five eyes) showed very poor visual acuity (2.4 Âą 0.46 logMAR, hand motion) at the final follow-up. There was no significant difference in visual acuity between patients with RCH with VHL disease and sporadic RCH.
In comparing clinical features, patients with RCH with VHL disease tended to be diagnosed earlier than patients with sporadic RCH (p = 0.067). Bilateral RCH was observed only in patients with VHL disease but not in patients with sporadic RCH. In terms of tumor location, juxtapapillary RCH was observed only in patients with VHL disease. There were no statistically significant differences between the two groups in terms of mean visual acuity, tumor size, type, and complications.
We performed treatment considering the size, location, and number of RCHs and the presence of associated complications such as exudative or tractional retinal detachment (Fig. 1). Seven eyes were observed without treatment. Ten eyes were treated with antivascular endothelial growth factor (anti-VEGF) agents, and five eyes were responsive to treatment (decreased subretinal fluid or exudation). Laser photocoagulation (focal or barrier) treatment was performed on the peripheral small lesions of eight eyes. Photodynamic therapy was performed on three eyes. Six eyes were treated with vitrectomy for exudative or tractional retinal detachment, ERM, or vitreous hemorrhage. Four eyes (67%) underwent more than two vitrectomies. During vitrectomy, silicone oil injection was performed in five eyes (83%). Five eyes were treated with cryotherapy, four of them were treated while performing vitrectomy. The treatment methods and the number of eyes requiring treatment are summarized in Table 2.
Discussion
The VHL protein is essential for the degradation of hypoxia-inducible factor-1Îą (HIF-1Îą) and HIF-2Îą. Loss of VHL leads to abnormal cell production and consequent abnormal production of proangiogenic factors such as VEGF, which are responsible for abnormal vascular changes and hemangioblastomas in the retina [18]. VHL is a hereditary multisystem syndrome; VHL incidence is approximately 1 in 36,000 live births and is inherited in an autosomal dominant pattern. This disease is characterized by the growth of various benign or malignant tumors of the retina and brain, along with cysts of several visceral organs, such as the kidneys, pancreas, adrenal glands, and reproductive organs [16].
Germline mutations in VHL show a diverse spectrum according to ethnic backgrounds. The nonsynonymous variant c.208G>A (p.Glu70Lys) in the VHL gene is frequently detected in Korean patients with VHL [14]. However, this variant has rarely been reported in the Western countries, even in Chinese and Japanese populations. Changes in codon 167 of the VHL gene are frequently observed in the Western and Japanese populations. P.Arg-167Trp, p.Arg167Gln, p.Ser65Leu, p.Ser65Trp, and p.Arg161Ter are mutational hot spots in the Chinese population [12,19]. Glu70Lys mutation is known to cause type 1 VHL disease, which is associated with central nervous system hemangioblastoma, retinal hemangioblastoma, and renal cell carcinoma, but not pheochromocytoma [20]. In our study, of the four genetically confirmed patients with VHL disease-related RCH, three patients had a mutation of Glu70Lys, and the other had a mutation of c.264G>A, p.Trp88Ter. Patients with the p.Trp88Ter mutation exhibit a type 2 phenotype of VHL disease.
The genotypic phenotypic correlation of RCH has been studied. One study showed that patients with a truncating variant developed RCH earlier and had higher age-related incidence than patients with a single amino acid substitution. The study proposed that patients with a truncating variant may be considered for greater intensive ophthalmological monitoring [21]. In another study, the genotype category was classified into and compared as three categories (amino acid substitutions, protein-truncating variants, and complete deletions of VHL protein). This study showed that patients with complete deletion had the lowest prevalence of RCH and were associated with the highest mean visual acuity compared to patients in the other two genotype categories [22]. In our study, three patients with the Glu70Lys mutation RCH appeared to have a mild clinical course, even though the sample size was too small to be demonstrated (Fig. 2A-2F). One patient with a p.Trp88 mutation showed bilateral progressive retinopathy and a type 2 VHL disease phenotype (with pheochromocytoma). We assumed that the nonsense mutation type might contribute to a relatively aggressive feature and VHL type; however, a previous study revealed that most type 2 VHL diseases were missense mutations [14].
Although family members may share the same causative gene, phenotypic features can manifest differently due to the phenotypic heterogeneity of VHL disease. In our cohort, one proband (Fig. 2A) with bilateral RCH showed no other systemic tumors, but her father had only paraganglioma without RCH, and her younger sibling had no tumors despite having the same causative gene. Therefore, it is suggested that those with a family history of VHL without any existing symptoms should undergo regular examinations according to the guidelines [9]. When it comes to RCH screening, dilated funduscopic examination should be performed every 12 months. Fluorescein angiography could be helpful in finding early or peripheral small lesions [1].
In a previous study comparing sporadic and VHL disease-related RCH, the median age at diagnosis was earlier in VHL-related RCH than sporadic RCH [23]. Although it was not statistically significant, our study also showed a similar tendency in terms of differences in age at diagnosis. Regarding the number of tumors, some of our solitary RCH patients have confirmed VHL disease. This finding has also been identified in other studies [15,24]. Therefore, even in patients with only solitary RCH and lacking other systemic or family history of VHL disease, the possibility of underlying VHL disease with its associated morbidity should be considered [15]. And consistent with previous studies, juxtapapillary RCH was only observed in patients with VHL disease. Therefore, the presence of juxtapapillary RCH (Fig. 1) strongly points towards VHL disease [25].
The visual outcome of patients with VHL is dependent on the size, location, and number of RCHs and the presence of associated findings such as exudative or tractional retinal detachment [23]. In our study, there was no definite correlation between tumor size, location, and visual prognosis. However, associated complications, specifically exudative and tractional retinal detachments, were definitively related to poor visual outcomes.
Depending on the tumor size, location, and complications, intravitreal anti-VEGF injection, laser photocoagulation, cryotherapy, and vitrectomy were selected for treatment. With laser photocoagulation, complete histopathological degeneration is possible in tumors with one-disc diameter, and clinical improvement can be achieved in tumors with disc diameters of up to 2.5-disc diameters (<4.5 mm) [26]. Large RCH in the peripheral retina, especially accompanying exudation, cryotherapy should be considered [27]. Pars plana vitrectomy is considered in cases with significantly decreased visual acuity caused by a refractory tumor or in the presence of an ERM, macular edema, and retinal detachment [28]. Previous studies showed relatively good visual outcomes, and a stable retinal state could be achieved with the proper intervention [28,29].
The limitation of this study is that there were not many patients because it is a rare disease. Because there were fewer patients who had genetic testing, it was difficult to compare differences according to genetic variations. Due to the small number of cases included, it was hard to find the genotype-phenotype correlations of RCH.
In conclusion, current advances in genetic diagnostic technology allow for the accurate identification of pathogenic variants of the VHL gene. It is important to understand the clinical characteristics of genetically confirmed VHL disease-related RCH for the surveillance and management of patients with VHL with or without RCH. Here, we report two genetic mutations in VHL disease-related RCH in the Korean population. This is the first report of the clinical features of VHL disease-related RCH in the Korean population. Through genetic diagnosis of RCH patients, we hope to offer better management of these patients.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print